
Real-Time PCR
In Real-Time PCR, when PCR is taking place then on a computer screen it can be seen that the product is being formed or not. There is no need to wait for the end product as the apparatus is connected with the computer.In conventional PCR, it takes more time. One has to wait till the end so the product can be run on gel parallel to the marker i.e DNA Standard.
The product of contamination will be dim. This is due to the fact that is more primers are designed according to the product so there can be less product of contamination. The hand of Amina products will be bright.

Why markers are used?
When the marker is run then the product of primer dimer can be cherished from other (original product). In the case of primer dimer formation, there is also land but this band is not the main product. If the lost nucleotide at 3'end of beta primers is hot compensatory then there will be no product as a result of dime markers.
In the case of dimer formation, some primers get wasted. If as a result of dimer formation, product forms then dNTPs also get wanted. If genome size is large then there will be more non-specific binding/product.
Complete binding of primer with the template is not necessary. Synthesis can occur only if 3' end binds.
The primer length in RAPD (Random Amplified Polymorphism DNA) is 7-8 nucleotides.
In order to get rid of non-specific product and to obtain just required product:
- Nested PCR can be used.
- The time given to enzyme can be reduced e.g if enzyme makes 1kb in 1 minute and the initial duration given to it is 2 minutes in which it makes products off 2kb or 1.5kb then by reducing the time given to the enzyme. For synthesis, the products like 2kb or 1.5kb will net form as will remain unfinished. The enzyme will not be able to complete the motion & will leave only fragments. Thus only the required product will form.
- Annealing temperature can also be increased. At high-temperature primer will only bind specifically & will have strong binding.
3' end should not be complementary.
Primer dimer product will be small in size & will come at a lower position. An experienced worker knew the position & brightness of the band & also knows the time for which gel is run & at what voltage. Thus, in such a case marker is not required. But in order to defend one’s work, a marker is required.
Advantages o Real-Time PCR
- Time-saving
- Easy
- Product formation is quantified online.
- Viral load i.e number of viruses can be detected.
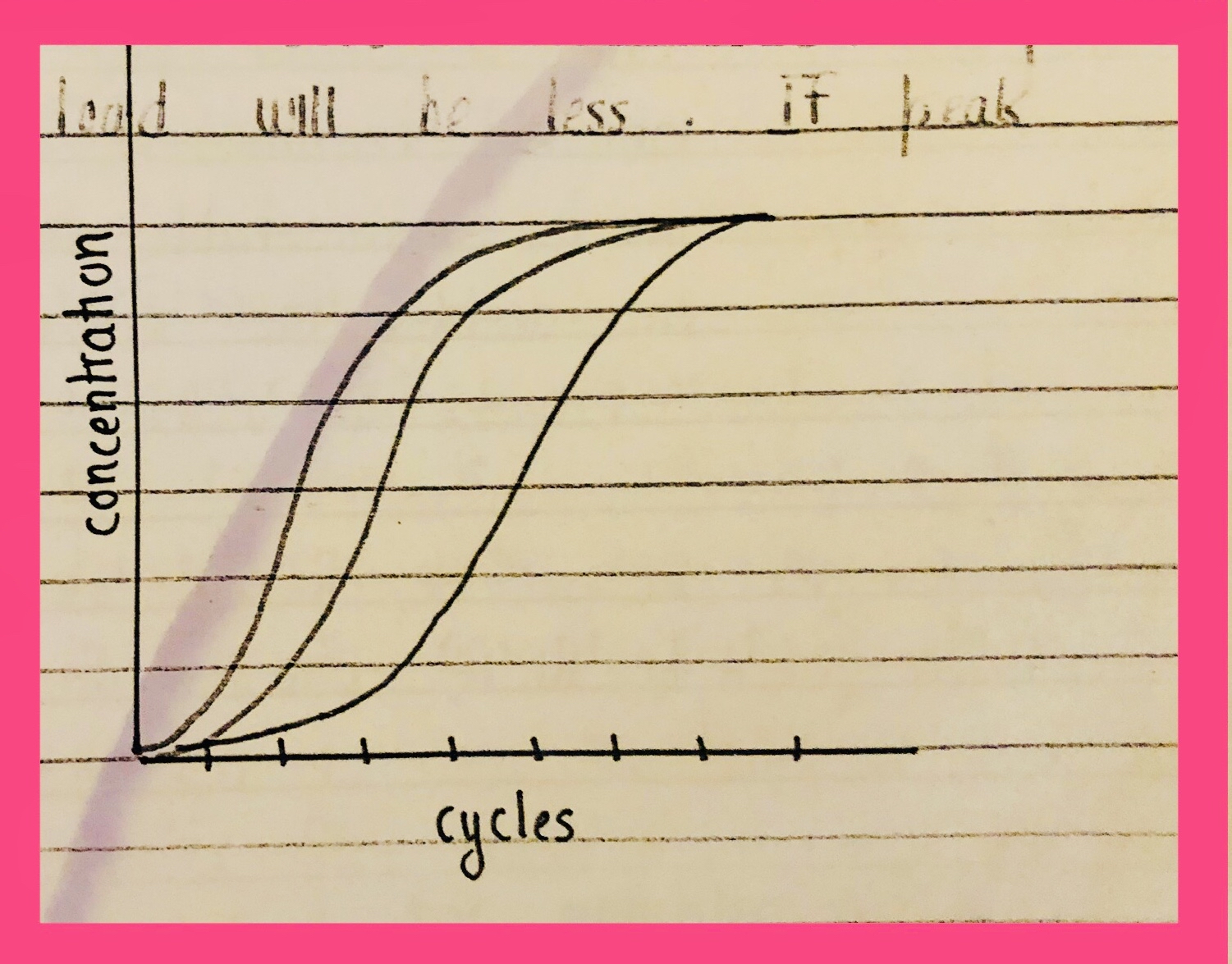
Treatment varies on the basis of the amount of virus. If the load of HIV is more then there will be AIDS otherwise not. Being HIV + & having AIDS are two different things.
More load ➡️ fewer T lymphocytes
Less load ➡️ more T lymphocytes
⬇️
The immune system will be working efficiently.
Components of real-time PCR and conventional PCR are the same then what is the difference?
What is the difference between the RT PCR &real time PCR?
RT PCR is reverse transcriptase PCR.
In RT PCR

Enzymes i.e DNase or RNase are used in order to make the simple (DNA or RNA) pore is to remove the other material. If one is sure of the purity of the sample then there is no used to treat with an enzyme.
Detection (sensitivity) & data interpretation of Real Time PCR & contential PCR differs.
The sensitivity of reagents varies in both PCRs. (more in Real-Time PCR)
More than 30 million copies from in 25-30 cycles.
The detection system used in real-time is of two types.
- Specific detection system.
- Non-specific detection system.
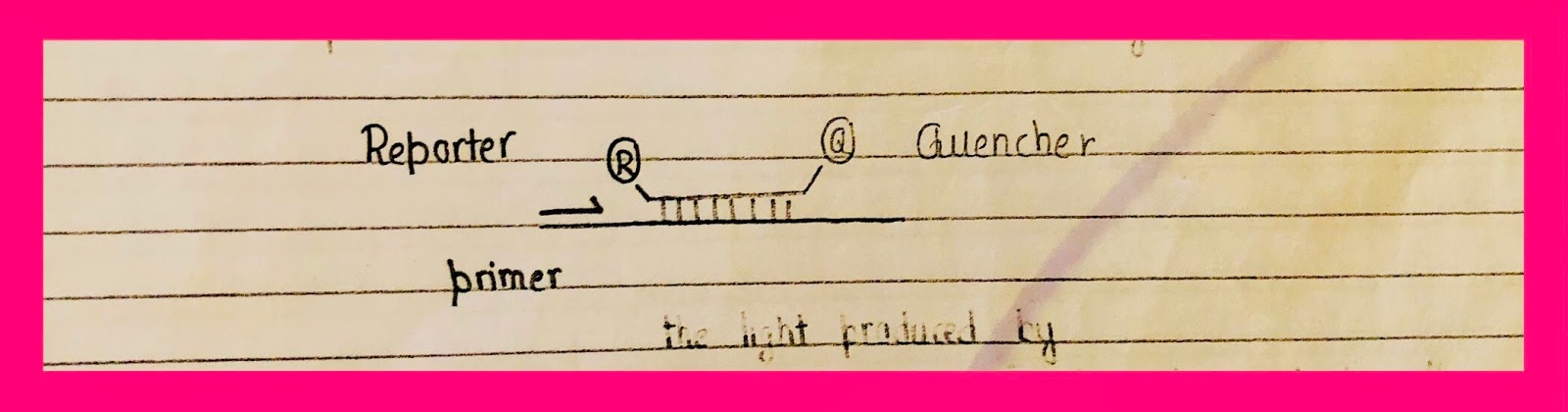
Specific Detection System ➡️ TaqMan assay/probe.
- 5' nuclease assay. Binds with template only when the sequence is complementary.
Non-specific Detection System ➡️ Syber green.
- Similar to ethidium bromide but moe sensitive (1000 times)
- Specific for double-stranded DNA (any double-stranded).
When present on the same strand reporter will be absorbed by the quencher. When the enzyme reaches the reporter, it is released & thus, emits light. This method of detection is more expensive as compared to syber green but it has the benefit that the TaqMan probe will only bind with specific sequence & fluorescence will increase any if primer binds with the sequence.
A TaqMan probe can also be used for multiplex qPCR. In this PCR, more than one product is amplified in a single tube using a single template. More than one gene can be amplified. As genes are different so primers will be different e.g
- One primer pair for gene A.
- One primer pair for gene B.
In multiplex PCR, products will be different sizes so we can tell that which product belongs to which gene e.g in the case of the TaqMan probe if two products then there will be two peaks. Fluorescent signals will so be different as dyes will be different e.g
Gene A ➡️ Green
Gene B ➡️ Red
Gene C ➡️ Blue/Yellow
The laser will read the signals. Taqman probe can be used in multiplex PCR provided that dyes will be different in fluorescence in order to differentiate to see different peaks in a different plateau.
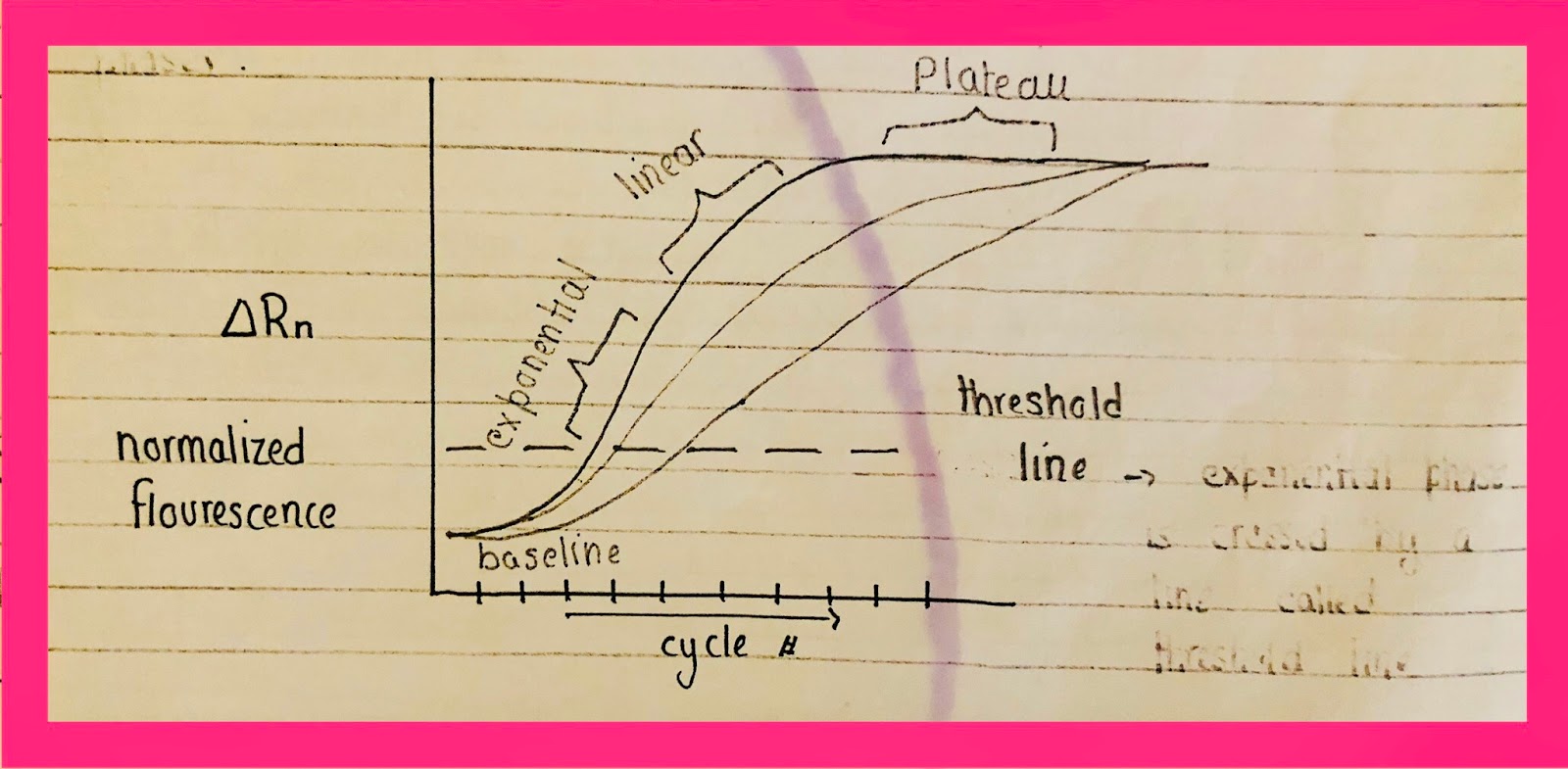
The threshold line is an arbitrary line with no exact position. It is usually drawn at a point when the exponential phase rises from the line. The baseline will form even when there is no synthesis. Reference dye will give color at baseline hence it will not start from zero. CDNA will be in less amount. It is a ground line and nothing can be concluded on its base.
The exponential phase is important for analysis &quantification. From the analytical point of view, plateau & linear are not important. The rise of the exponential phase shows product formation. Right from its beginning readable signal can be obtained. After 10-15 cycles, if no signal appears or enzyme falls short. The dye can also lesser in amount. Template increases in number. Therefore, analysis is carried from the threshold line when there are no such complications.
The starting point in Real-Time PCR or qPCR is DNA whereas in RT PCR it is mRNA.
Quantify means to analyze data. There are two methods for this:
- Absolute Method ➡️ with the help of a standard curve.
- Relative Method or Comparative Ct Method.
In Absolute Method, we calculate the number of template molecules or products or the number of starting molecules with the help of a standard curve.
The threshold line is not baseline. The threshold line starts after the baseline when the exponential phase has started.
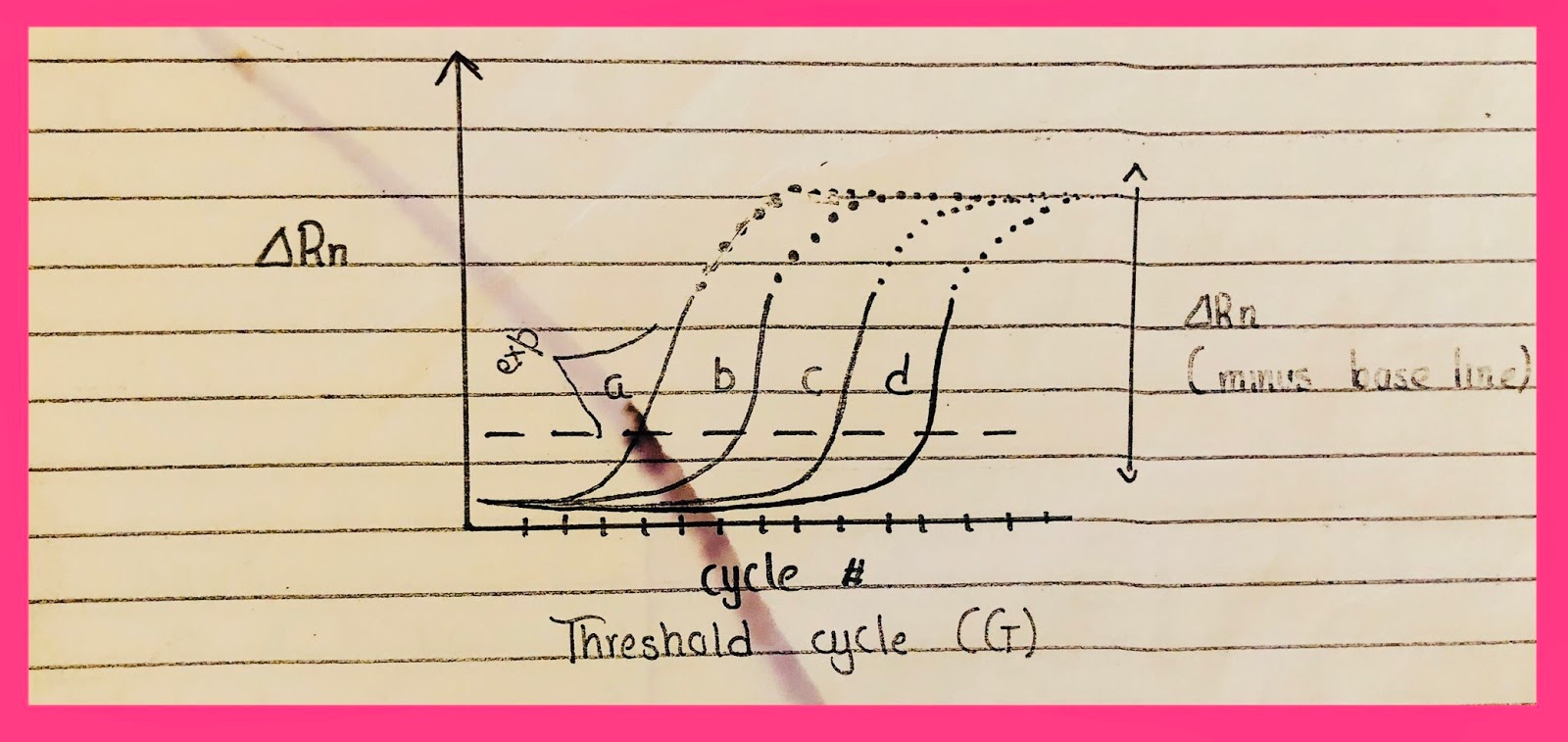
Ct is the amount of DNA which crosses the threshold line. The cycle number of the point of cross of the threshold line with the exponential line is Ct (Ct cross threshold line).
The fluorescence coming from the dye is Rn. When the baseline is substracted then it is Rn.
Ct has an inverse relation with DNA.
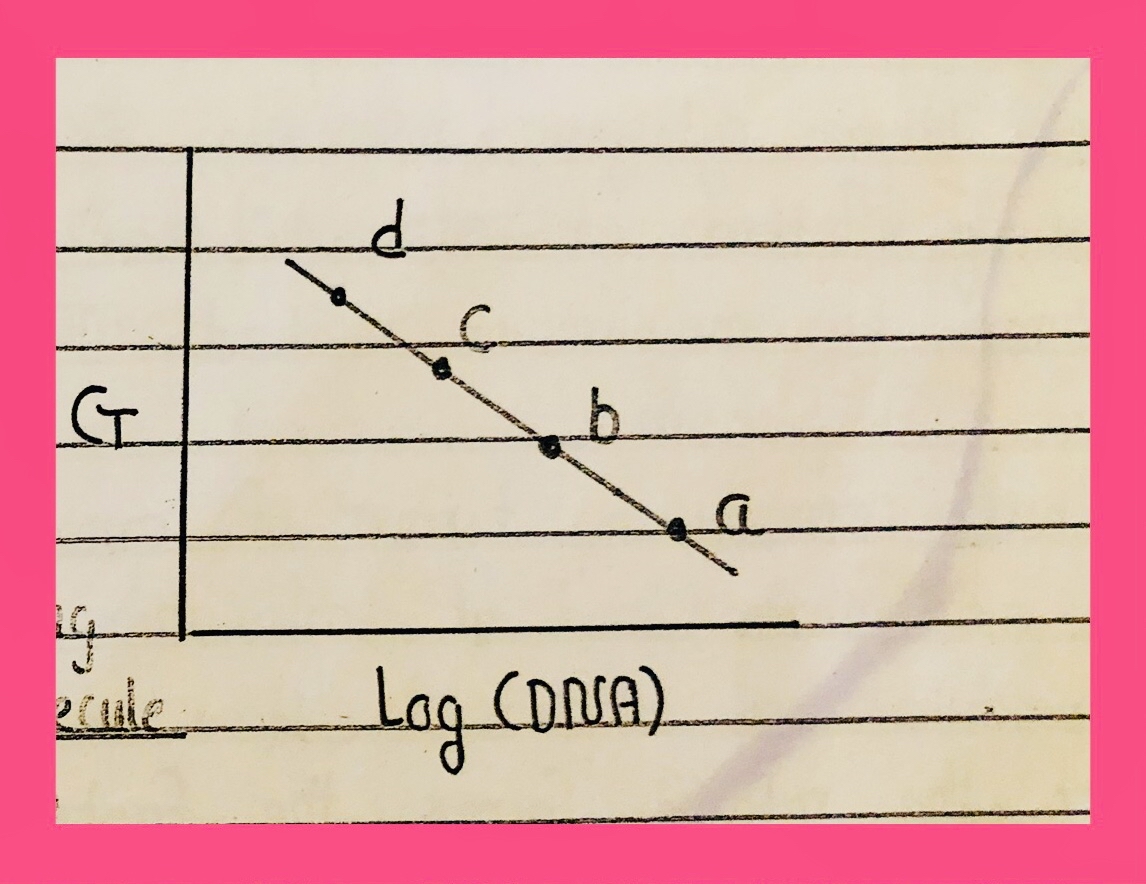
Dilutions of a sample are run e.g 10ng, 5ng, 2.5ng, 1.25ng, 0.6ng. As the amount of template molecule reduces so more delayed will be the expression/exponential phase i.e rise after a long time. The crossing of the threshold line will take more time i.e after more cycles. Then the sample to be tested is run and its rise is compared to that of the standard curve.
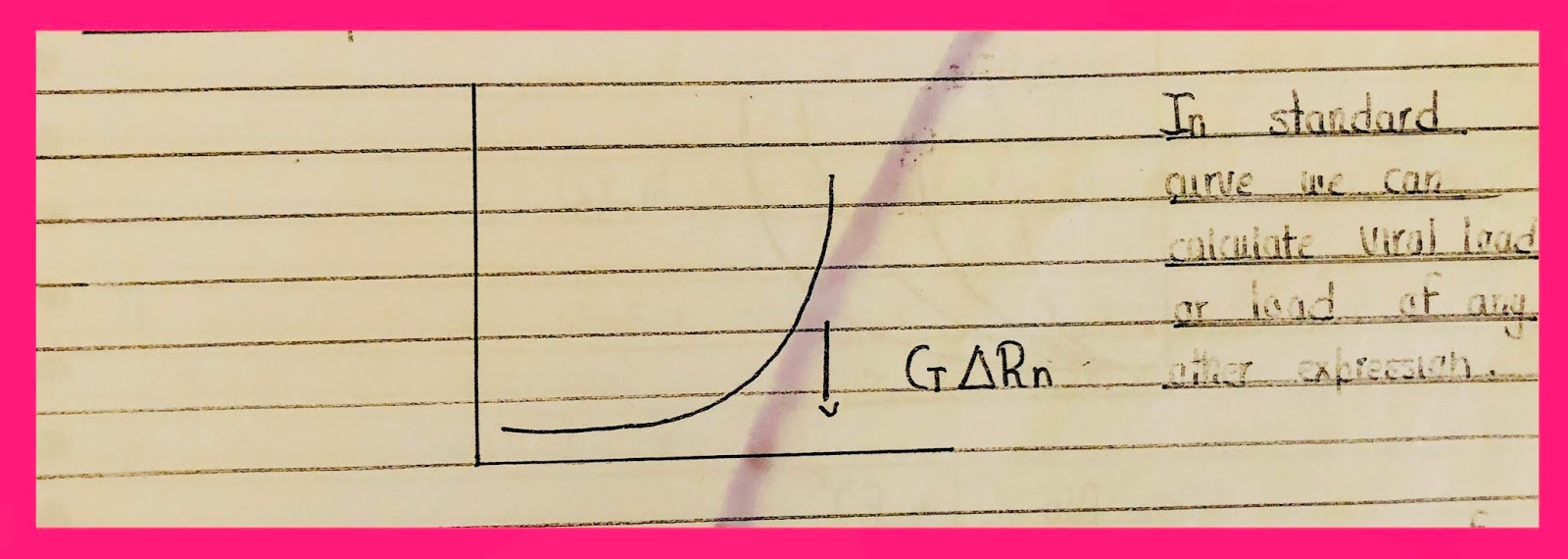
In the standard cure, we can calculate viral lead or load of any other expression.
In an absolute method, the standard curve is required but in the case of the comparative method, the standard curve is not required. In the comparative method, there are two genes, one is experimental, and the other is control. Within a system, there will be a control gene.
The equation is employed and the amount of experiment or its fold is obtained. For expression studies, if gene expression increase in one case so one sample i.e control will be untreated and others will be treated. The basic use of Ct expression is to see the difference in the expression of genes in treated and untreated cases.
When cytokine, hormone, or drug is being used or there is an infection or disease then the expression of a gene can be analyzed by the comparative method. There is no need for a standard curve. A comparison is carried simply with control.
Comparison is not carried in terms of concentration or copy number but fold. At the end of the calculation, it is calculated how much fold has increased.
How much mRNA has formed?
The more mRNA forms the faster the peak will enter into the exponential phase. If the amount of template mRNA is less then the rise in peak will take more time and if there is no expression then there will be no rise in time.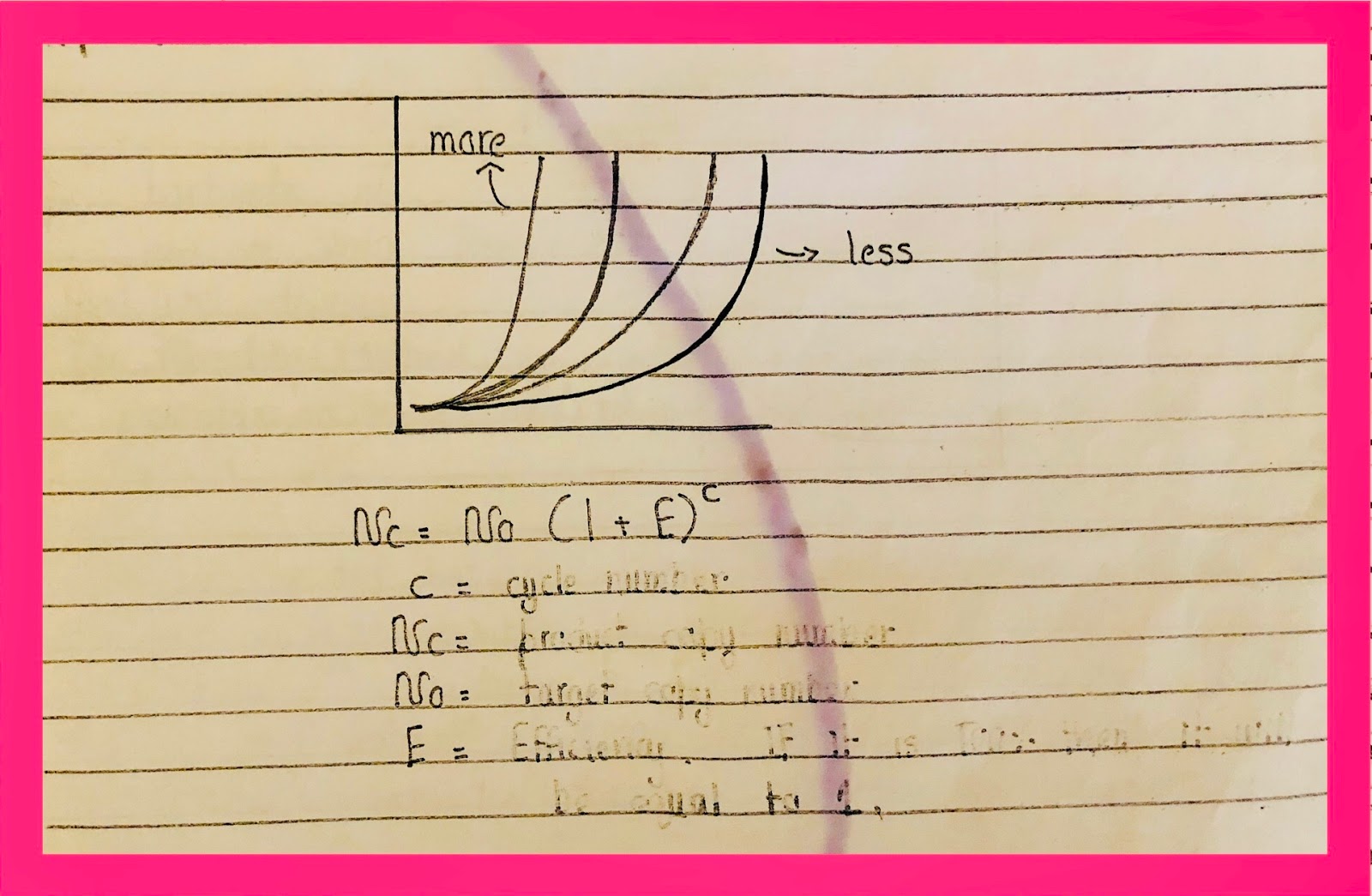
- Efficiency is 100% when the line is in the exponential phase.
- Efficiency cannot be 100% in linear or plateau.
Equation of comparative method:
Fold increase (Increase) = 2^-🛆🛆Ct
(Answer should not be negative i.e in minus)
Sample A (treated or untreated) is subtracted from sample B (treated or untreated).
2^-🛆🛆Ct = Sample A - Sample B
= (Ct of the gene of interest - Ct of internal control)
-
(Ct of the gene of interest - Ct of internal control)
It depends on us whether where we put treated and where untreated. In one case both will be treated while in other cases both will be untreated e.g
HOXD 10 is a gene whose expression is being studied.
Treated Ct value = 24.6
Untreated Ct value = 27.5
18s RNA ➡️ Treated = 9.9 Untreated = 9.8
= 2^-(24.6-9.9) - (27.5-9.8)
= 2 ^ 14.7 - 17.7
=2^-(-3) = 2^ 3 = 8 fold incease
An increase in expression level causes a decrease in Ct value.
Applications of Real-Time PCR
- Diagnostic Tool: To detect the presence of viruses or bacteria present in a sample of tissue from any animal or plant. Known primers will be designed against it. The sequence should be specific to that organism it should not be present in the human sequence. In order to confirm the specie, set or pairs of primers will be used against that bacteria or virus. With the help of real-time PCR, the genome of that bacteria or virus can be amplified.
- To quantify gene expression
- To calculate the number of viruses/bacteria present per volume.
- To see the extent of transcription of a gene more expression ➡️ more mRNA produces ➡️ more transcription.
- Lessor no mRNA ➡️ low or no expression.
- Difference between diseased & normal genes.
- Change in gene expression in relation to differentiation.
- Change due to exposure
One sample will be unexposed and others will be exposed. In this way, change can be observed. E.g giving a dose of insulin will result in low expression of insulin gene as it will be already present.
- To study non-coding RNA expression.
- To validate small interfering RNA.
- Micro-organism Dectection.
A genome sequence is known
⬇️
Primer is designed
⬇️
Product size is known
⬇️
Conventional PCR Genome amplified
Editor's Recommendation:
- Analysing Metabolic Pathways
- Protein Threading Sequence
- Ab Initio Protein Structure Prediction
- Homology Modeling
- Hot Start PCR, Multiplex PCR, Avoiding Contamination In PCR, Advantages, and Disadvantages in PCR
- DNA Damage
- Docking | Protein-Protein Docking | Protein-Ligand Docking
- Functional Regulation | Genetic Aspect | Indirect Aspects
- Database Development
- Functional Analysis At Structure Level
- PTMs and Functional Regulations
- Modeling Cellular Processes
- PCR Reagents | Stochastic Effect | STR Classification
- DNA Degradation
- DNA Quantification | Human DNA Quantification Method | Advantages
- Desirable Characteristics of STR used in Forensic DNA typing
- DNA Ladders
- Metabolic Pathways
- Non-Human DNA
- Mitochondrial DNA
- Integrated Genomic Circuits
- Shutter Product Formation
- Mini STR Sites
- Immuno Quantitative Assay
- Molecular Diagnosis of Genetic Diseases
- STR Sites
Real-Time PCR
 Reviewed by Abdullah
on
June 24, 2020
Rating:
Reviewed by Abdullah
on
June 24, 2020
Rating:
Reviewed by Abdullah
on
June 24, 2020
Rating:
No comments:
Don't add any Spam link in comment box.